Unsupervised Learning I - Clustering Analysis
Patrick PUN Chi Seng (NTU Sg)
References
Chapter 12.5.3, [ISLR2] An Introduction to Statistical Learning - with Applications in R (2nd Edition). Free access to download the book: https://www.statlearning.com/
To see the help file of a function funcname, type
?funcname.
K-Means Clustering
The function kmeans() performs K-means clustering in
R. We begin with a simple simulated example in which there
truly are two clusters in the data: the first 25 observations have a
mean shift relative to the next 25 observations.
set.seed(1)
x <- matrix(rnorm(50 * 2), ncol = 2)
x[1:25, 1] <- x[1:25, 1] + 3
x[1:25, 2] <- x[1:25, 2] - 4We now perform K-means clustering with K=2.
km.out <- kmeans(x, 2, nstart = 20)The cluster assignments of the 50 observations are contained in
km.out$cluster.
km.out$cluster## [1] 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 2 2 2 2 2 2 2 2 2 2 2 2 2
## [39] 2 2 2 2 2 2 2 2 2 2 2 2The K-means clustering perfectly separated the observations into two
clusters even though we did not supply any group information to
kmeans(). We can plot the data, with each observation
colored according to its cluster assignment.
#par(mfrow = c(1, 2))
plot(x, col = (km.out$cluster + 1),
main = "K-Means Clustering Results with K = 2",
xlab = "", ylab = "", pch = 20, cex = 2)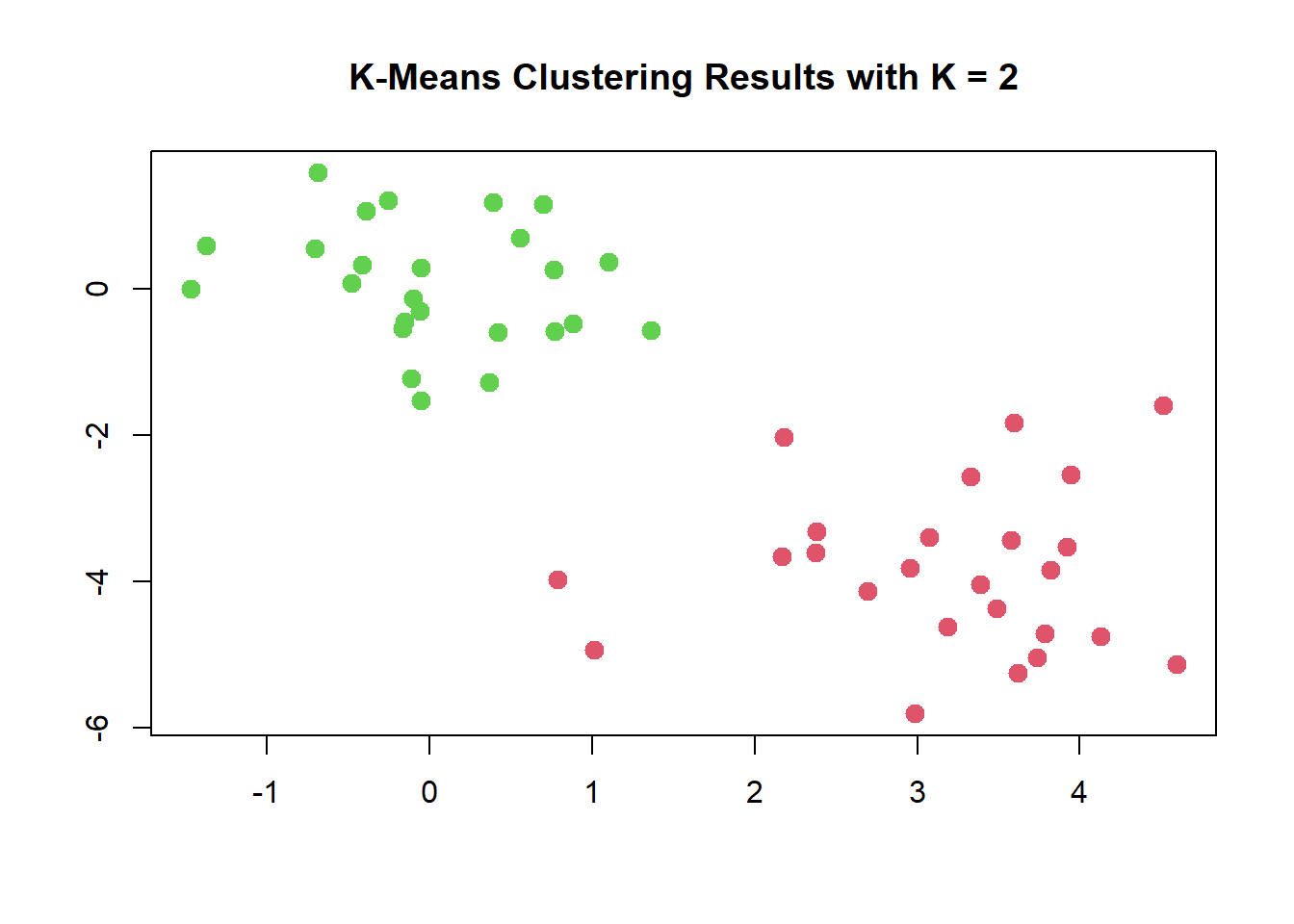
In this example, we knew that there really were two clusters because we generated the data. However, for real data, in general we do not know the true number of clusters. We could instead have performed K-means clustering on this example with K=3.
set.seed(2)
km.out <- kmeans(x, 3, nstart = 20)
km.out## K-means clustering with 3 clusters of sizes 20, 25, 5
##
## Cluster means:
## [,1] [,2]
## 1 3.08290361 -4.26589906
## 2 0.03223135 0.06924384
## 3 3.51171162 -2.10935842
##
## Clustering vector:
## [1] 1 1 1 1 3 3 1 1 1 1 3 1 1 1 1 1 1 3 1 3 1 1 1 1 1 2 2 2 2 2 2 2 2 2 2 2 2 2
## [39] 2 2 2 2 2 2 2 2 2 2 2 2
##
## Within cluster sum of squares by cluster:
## [1] 27.901401 28.534174 3.740301
## (between_SS / total_SS = 84.7 %)
##
## Available components:
##
## [1] "cluster" "centers" "totss" "withinss" "tot.withinss"
## [6] "betweenss" "size" "iter" "ifault"plot(x, col = (km.out$cluster + 1),
main = "K-Means Clustering Results with K = 3",
xlab = "", ylab = "", pch = 20, cex = 2)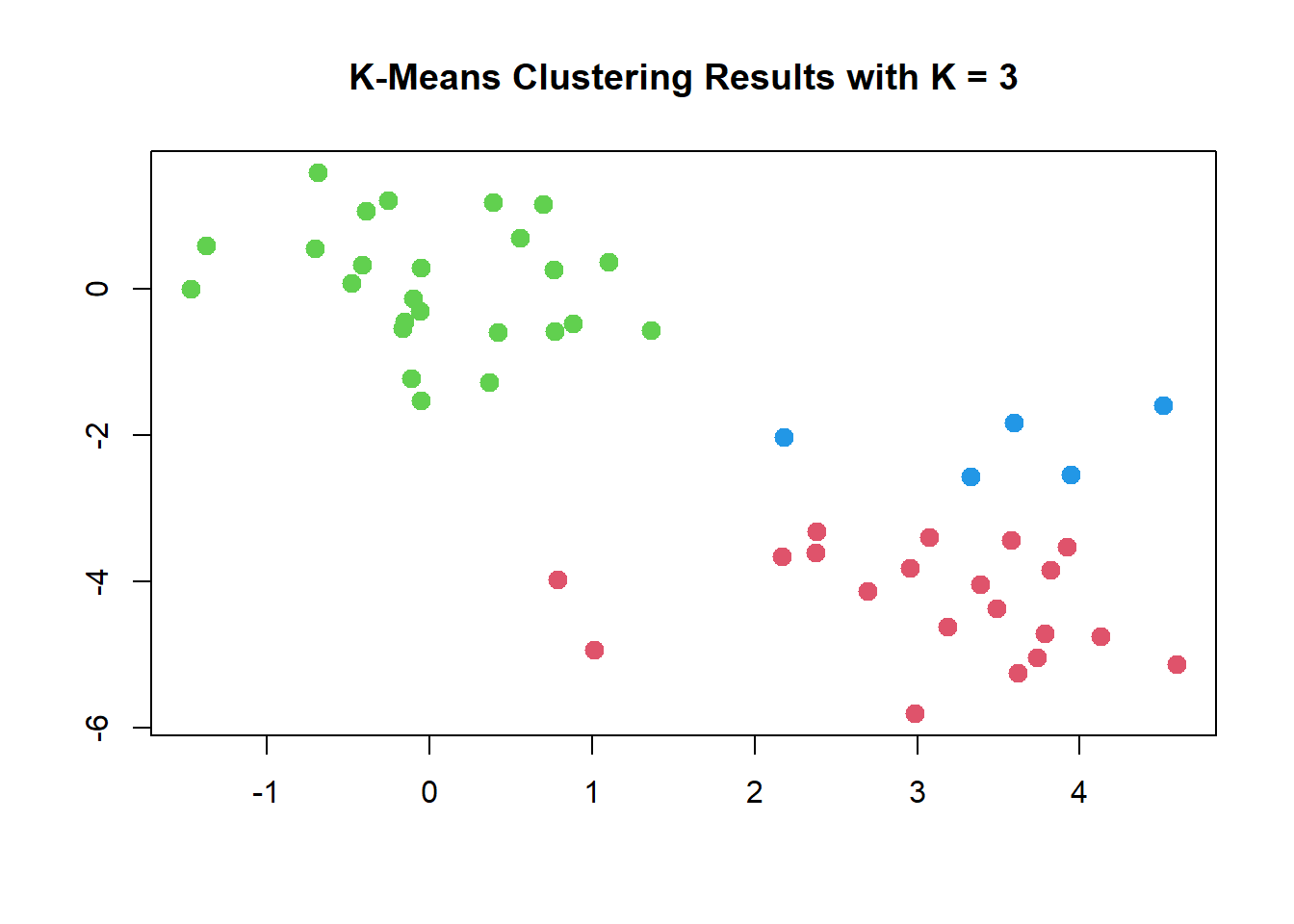
When K=3, K-means clustering splits up the two clusters.
To run the kmeans() function in R with
multiple initial cluster assignments, we use the nstart
argument. If a value of nstart greater than one is used,
then K-means clustering will be performed using multiple random
assignments in Step 1 of K-means Clustering Algorithm, and the
kmeans() function will report only the best results. Here
we compare using nstart = 1 to
nstart = 20.
set.seed(3)
km.out <- kmeans(x, 3, nstart = 1)
km.out$tot.withinss## [1] 60.17588km.out <- kmeans(x, 3, nstart = 20)
km.out$tot.withinss## [1] 60.17588Note that km.out$tot.withinss is the total
within-cluster sum of squares, which we seek to minimize by performing
K-means clustering. The individual within-cluster sum-of-squares are
contained in the vector km.out$withinss.
We strongly recommend always running K-means clustering with
a large value of nstart, such as 20 or 50, since otherwise
an undesirable local optimum may be obtained.
When performing K-means clustering, in addition to using multiple
initial cluster assignments, it is also important to set a random seed
using the set.seed() function. This way, the initial
cluster assignments in Step 1 can be replicated, and the K-means output
will be fully reproducible.
Hierarchical Clustering
The hclust() function implements hierarchical clustering
in R. In the following example we use the simulated data
before to plot the hierarchical clustering dendrogram using complete,
single, and average linkage clustering, with Euclidean distance as the
dissimilarity measure.
We begin by clustering observations using complete linkage. The
dist() function is used to compute the \(50 \times 50\) inter-observation Euclidean
distance matrix.
hc.complete <- hclust(dist(x), method = "complete")We could just as easily perform hierarchical clustering with average or single linkage instead:
hc.average <- hclust(dist(x), method = "average")
hc.single <- hclust(dist(x), method = "single")We can now plot the dendrograms obtained using the usual
plot() function. The numbers at the bottom of the plot
identify each observation.
par(mfrow = c(1, 3))
plot(hc.complete, main = "Complete Linkage",
xlab = "", sub = "", cex = .9)
plot(hc.average, main = "Average Linkage",
xlab = "", sub = "", cex = .9)
plot(hc.single, main = "Single Linkage",
xlab = "", sub = "", cex = .9)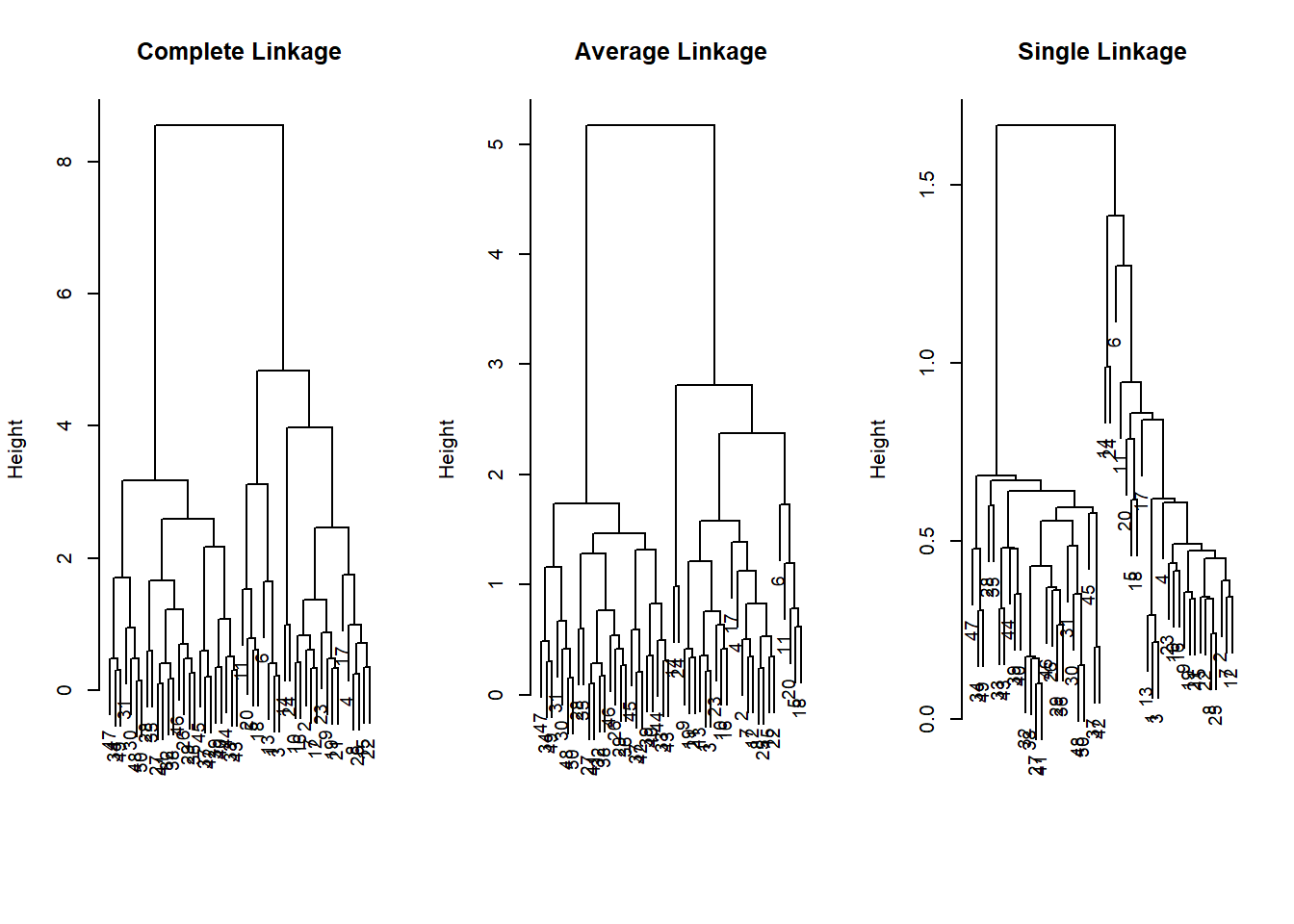
To determine the cluster labels for each observation associated with
a given cut of the dendrogram, we can use the cutree()
function:
cutree(hc.complete, 2)## [1] 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 2 2 2 2 2 2 2 2 2 2 2 2 2
## [39] 2 2 2 2 2 2 2 2 2 2 2 2cutree(hc.average, 2)## [1] 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 2 2 2 2 2 2 2 2 2 2 2 2 2
## [39] 2 2 2 2 2 2 2 2 2 2 2 2cutree(hc.single, 2)## [1] 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 2 2 2 2 2 2 2 2 2 2 2 2 2
## [39] 2 2 2 2 2 2 2 2 2 2 2 2The second argument to cutree() is the number of
clusters we wish to obtain.
For this data, complete and average linkage generally separate the observations into their correct groups. However, single linkage identifies one point as belonging to its own cluster. A more sensible answer is obtained when four clusters are selected, although there are still two singletons.
cutree(hc.single, 4)## [1] 1 1 1 1 1 2 1 1 1 1 1 1 1 3 1 1 1 1 1 1 1 1 1 3 1 4 4 4 4 4 4 4 4 4 4 4 4 4
## [39] 4 4 4 4 4 4 4 4 4 4 4 4To scale the variables before performing hierarchical clustering of
the observations, we use the scale() function:
xsc <- scale(x)
plot(hclust(dist(xsc), method = "complete"),
main = "Hierarchical Clustering with Scaled Features")
Correlation-based distance can be computed using the
as.dist() function, which converts an arbitrary square
symmetric matrix into a form that the hclust() function
recognizes as a distance matrix. However, this only makes sense for data
with at least three features since the absolute correlation between any
two observations with measurements on two features is always 1. Hence,
we will cluster a three-dimensional data set. This data set does not
contain any true clusters.
x <- matrix(rnorm(30 * 3), ncol = 3)
dd <- as.dist(sqrt((1 - cor(t(x)))/2))
plot(hclust(dd, method = "complete"),
main = "Complete Linkage with Correlation-Based Distance",
xlab = "", sub = "")
NCI60 Data Example
Unsupervised techniques are often used in the analysis of genomic
data. In particular, clustering and PCA are popular tools. We illustrate
these techniques on the NCI cancer cell line microarray
data, which consists of 6,830 gene expression measurements on 64 cancer
cell lines.
library(ISLR2)
nci.labs <- NCI60$labs
nci.data <- NCI60$dataEach cell line is labeled with a cancer type, given in
nci.labs. We do not make use of the cancer types in
performing clustering/PCA, as these are unsupervised techniques. But
after performing clustering/PCA, we will check to see the extent to
which these cancer types agree with the results of these unsupervised
techniques.
The data has 64 rows and 6,830 columns.
dim(nci.data)## [1] 64 6830We begin by examining the cancer types for the cell lines.
nci.labs[1:4]## [1] "CNS" "CNS" "CNS" "RENAL"table(nci.labs)## nci.labs
## BREAST CNS COLON K562A-repro K562B-repro LEUKEMIA
## 7 5 7 1 1 6
## MCF7A-repro MCF7D-repro MELANOMA NSCLC OVARIAN PROSTATE
## 1 1 8 9 6 2
## RENAL UNKNOWN
## 9 1Clustering the Observations of the NCI60 Data
We now proceed to hierarchically cluster the cell lines in the
NCI data, with the goal of finding out whether or not the
observations cluster into distinct types of cancer. To begin, we
standardize the variables to have mean zero and standard deviation one.
As mentioned earlier, this step is optional and should be performed only
if we want each gene to be on the same scale.
sd.data <- scale(nci.data)We now perform hierarchical clustering of the observations using complete, single, and average linkage. Euclidean distance is used as the dissimilarity measure.
par(mfrow = c(1, 3))
data.dist <- dist(sd.data)
plot(hclust(data.dist), xlab = "", sub = "", ylab = "",
labels = nci.labs, main = "Complete Linkage")
plot(hclust(data.dist, method = "average"),
labels = nci.labs, main = "Average Linkage",
xlab = "", sub = "", ylab = "")
plot(hclust(data.dist, method = "single"),
labels = nci.labs, main = "Single Linkage",
xlab = "", sub = "", ylab = "")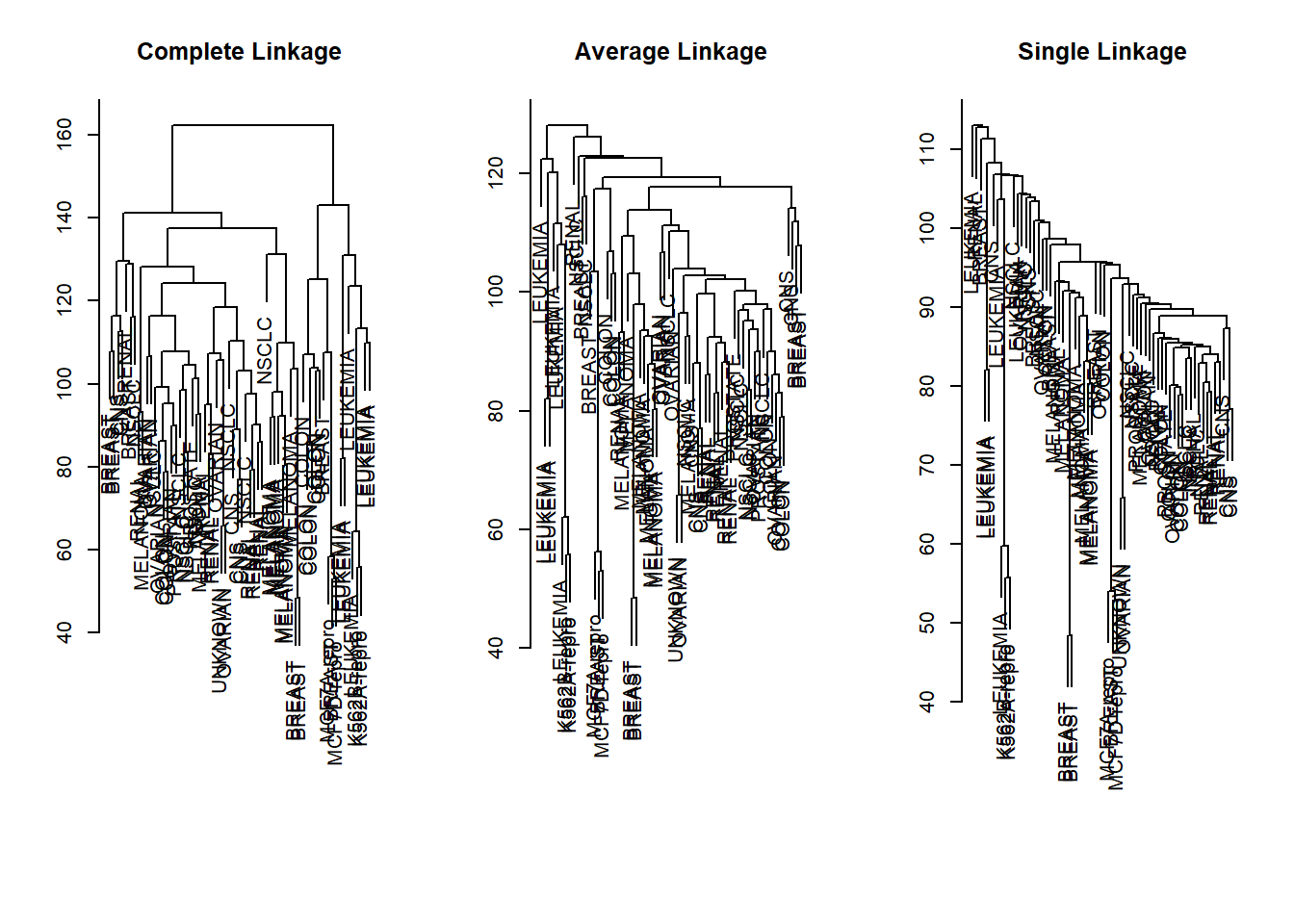
We see that the choice of linkage certainly does affect the results obtained. Typically, single linkage will tend to yield trailing clusters: very large clusters onto which individual observations attach one-by-one. On the other hand, complete and average linkage tend to yield more balanced, attractive clusters. For this reason, complete and average linkage are generally preferred to single linkage. Clearly cell lines within a single cancer type do tend to cluster together, although the clustering is not perfect. We will use complete linkage hierarchical clustering for the analysis that follows.
We can cut the dendrogram at the height that will yield a particular number of clusters, say four:
hc.out <- hclust(dist(sd.data))
hc.clusters <- cutree(hc.out, 4)
table(hc.clusters, nci.labs)## nci.labs
## hc.clusters BREAST CNS COLON K562A-repro K562B-repro LEUKEMIA MCF7A-repro
## 1 2 3 2 0 0 0 0
## 2 3 2 0 0 0 0 0
## 3 0 0 0 1 1 6 0
## 4 2 0 5 0 0 0 1
## nci.labs
## hc.clusters MCF7D-repro MELANOMA NSCLC OVARIAN PROSTATE RENAL UNKNOWN
## 1 0 8 8 6 2 8 1
## 2 0 0 1 0 0 1 0
## 3 0 0 0 0 0 0 0
## 4 1 0 0 0 0 0 0There are some clear patterns. All the leukemia cell lines fall in cluster 3, while the breast cancer cell lines are spread out over three different clusters. We can plot the cut on the dendrogram that produces these four clusters:
par(mfrow = c(1, 1))
plot(hc.out, labels = nci.labs)
abline(h = 139, col = "red")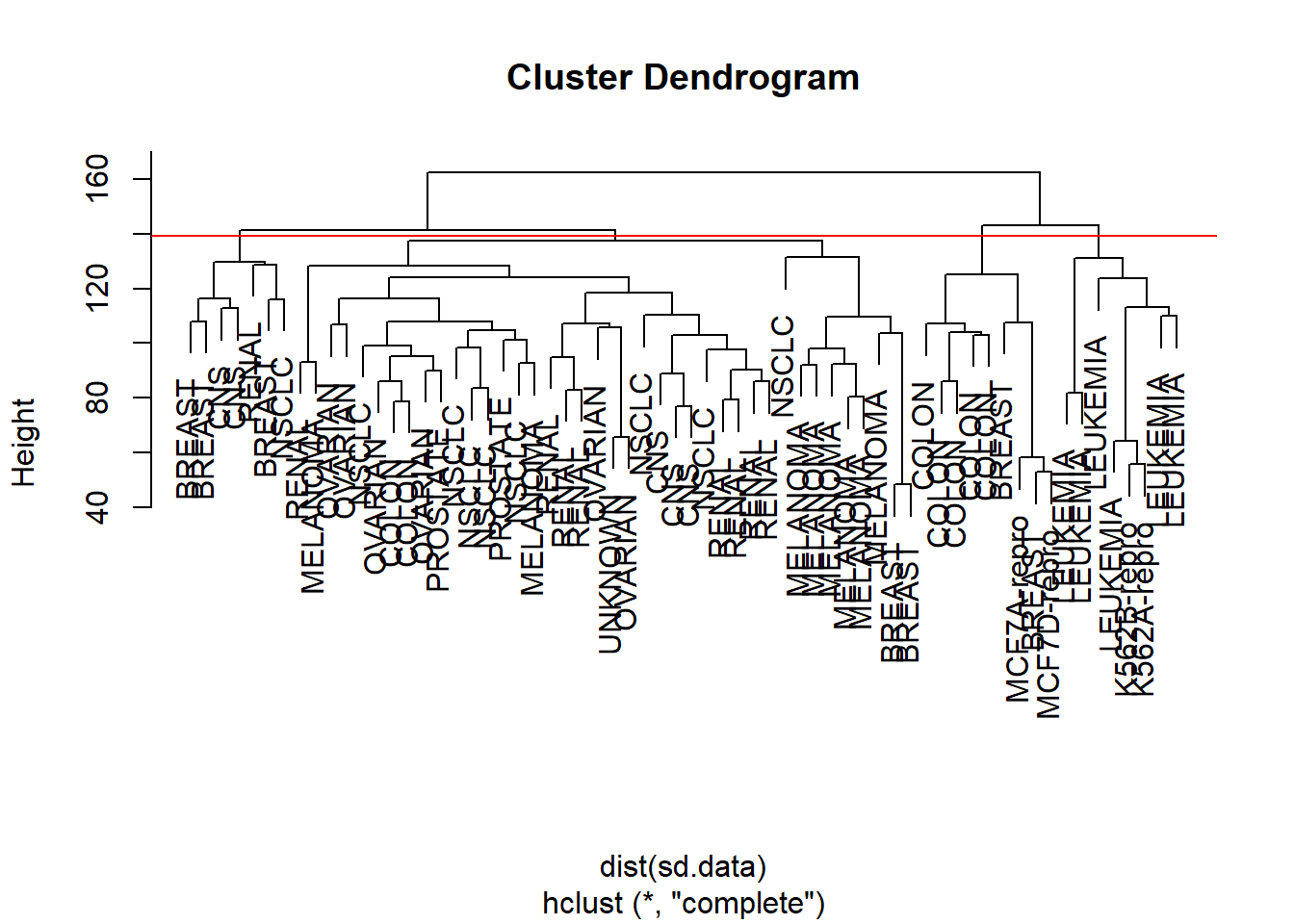
The abline() function draws a straight line on top of
any existing plot in R. The argument h = 139
plots a horizontal line at height 139 on the dendrogram; this is the
height that results in four distinct clusters. It is easy to verify that
the resulting clusters are the same as the ones we obtained using
cutree(hc.out, 4).
Printing the output of hclust gives a useful brief
summary of the object:
hc.out##
## Call:
## hclust(d = dist(sd.data))
##
## Cluster method : complete
## Distance : euclidean
## Number of objects: 64We claimed earlier that K-means clustering and hierarchical
clustering with the dendrogram cut to obtain the same number of clusters
can yield very different results. How do these NCI
hierarchical clustering results compare to what we get if we perform
K-means clustering with K=4?
set.seed(2)
km.out <- kmeans(sd.data, 4, nstart = 20)
km.clusters <- km.out$cluster
table(km.clusters, hc.clusters)## hc.clusters
## km.clusters 1 2 3 4
## 1 11 0 0 9
## 2 20 7 0 0
## 3 9 0 0 0
## 4 0 0 8 0We see that the four clusters obtained using hierarchical clustering and K-means clustering are somewhat different. Cluster 4 in K-means clustering is identical to cluster 3 in hierarchical clustering. However, the other clusters differ: for instance, cluster 2 in K-means clustering contains a portion of the observations assigned to cluster 1 by hierarchical clustering, as well as all of the observations assigned to cluster 2 by hierarchical clustering.