LU YUNPENG
Lecturer, Division of Chemistry and Biological Chemistry

|
Education: |
Ph.D.,
National University of Singapore |
|
Phone: |
(+65)
6513 2747 |
|
Email: |
|
|
Office: |
SPMS-CBC-06-23 |
Research Interests
Machine Learning in Chemistry
Recently, artificial intelligence
(AI), particularly in the form of machine learning (ML), has been largely
integrated into research works in cheminformatics and bioinformatics. ML is a branch
of AI which utilizes algorithms and mathematical models to train machines to
enhance their performance in assigned taskings. My research in this field is to
develop quantitative structure-activity relationship (QSAR) and quantitative
structure-property relationship (QSPR) based on machine learning models and
chemical data from database.
|
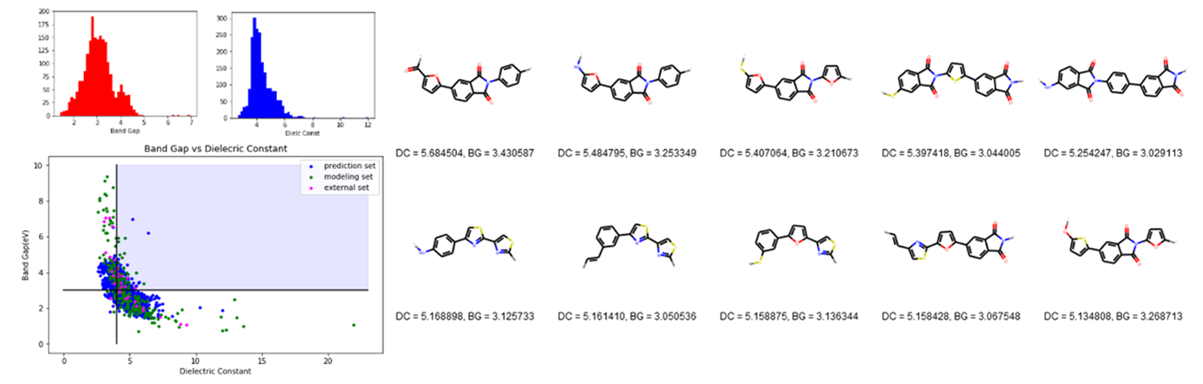
Figure 1. a) Histogram of band gap (top left) and b)
dielectric constant (top center) of newly generated polymers; c) Plot of band
gap against dielectric constant of the newly generated polymers together with
the polymers (bottom); d) the top 10 candidates with adequate band gap in the
combined dataset, ranked by dielectric constant value, where BG = Band Gap
(in eV) and DC = Dielectric Constant (right). |
DFT Calculations to Study Reaction Mechanisms
Understanding reaction mechanism
for a catalyzed chemical process is valuable to interpret reaction yields and
helps to optimize reaction conditions for future improvement. Density Functional
Theory (DFT) is the current “state-of-the-art” method to study reaction
mechanisms because of its balanced accuracy and computation efficiency. Over
years, we have collaborated with different synthetic chemistry research groups
in CBC to study reaction mechanism by DFT calculations.
|
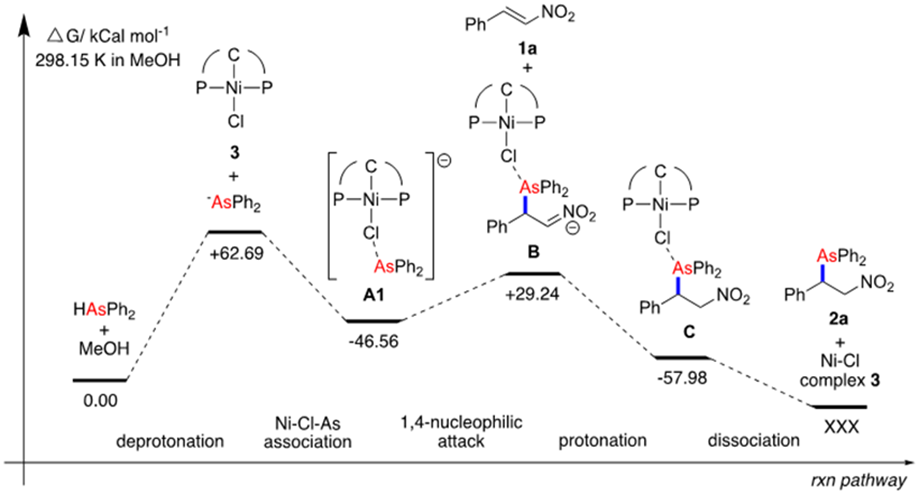
Figure 2. DFT calculations of the reaction profile for the
Ni(II)-mediated hydroarsination reaction at 298.15
K in MeOH |
Molecular Reaction Dynamics
Reactions are at the center of chemistry
both in chemistry laboratory experiments and theoretical studies. Molecular
reaction dynamics unfolds the history of chemical change on the molecular
level. It asks questions on what happens on the atomic length and time scales
as the chemical change occurs. Molecular reaction dynamics has become an
integral part of modern chemistry and is set to become a cornerstone for much
of the natural sciences. Theoretical work on polyatomic reactions dynamics
includes new potential energy surface calculations, direct dynamics studies,
calculation of isotope effects, and new approximated quantum scattering
methods. From 2011, we have published 9 peer-reviewed journal papers in this
field in the main stream journals, such as the Journal of Chemical Physics and
Physical Chemistry Chemical Physics.
|
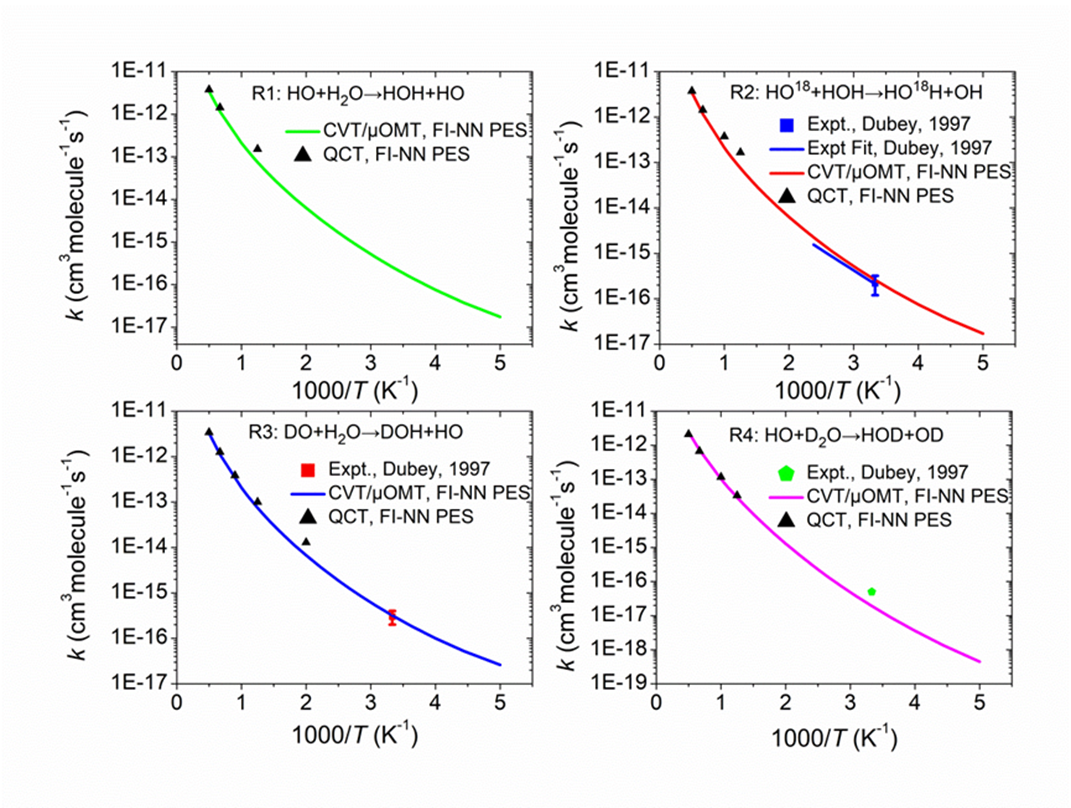
Figure 3. Calculated thermal rate coefficients for the
reaction OH + H2O → H2O + HO and its isotopologues in comparison to available experimental
values at T = 300 K. |
Selected Publications
1. Tay, WS; Lu, YP; Yang, XY; Li, YX; Pullarkat, SA; Leung, PH; “Catalytic and
Mechanistic Developments of the Nickel(II) Pincer
Complex-Catalyzed Hydroarsination Reaction”, CHEMISTRY-A
EUROPEAN JOURNAL, 25, 11308, (2019)
2. Ho, XL; Shao, HY; Ng, YY; Ganguly, R; Lu, YP;
Soo, HS, Visible Light Driven Hydrogen Evolution by Molecular Nickel Catalysts
with Time-Resolved Spectroscopic and DFT Insights, INORGANIC CHEMISTRY, 58, 1469,
(2019)
3. Zhu, YF; Lu, YP*; Song, HW; “Thermal rate coefficients and kinetic isotope
effects of the reaction HO + H2O → H2O + OH”, THEORETICAL
CHEMISTRY ACCOUNTS, 138, 111, (2019)
4. Song, HW; Lee, SY; Yang, MH; Lu, YP*, Six-dimensional and seven-dimensional
quantum dynamics study of the OH + CH4 → H2O + CH3
reaction, JOURNAL OF CHEMICAL PHYSICS, 139, 154310, (2013)
5. Song, HW; Lee, SY; Yang, MH; Lu, YP*, Full-dimensional quantum calculations
of the vibrational states of H5+, JOURNAL OF CHEMICAL PHYSICS, 138, 124309,
(2013)